NOTICE: this post was replaced by a newer version 13 March 2020. I will leave this post here, but strongly suggest you follow the new version instead.
I'm a big fan of using R for my MVPA, and have become an even bigger fan over the last year because of knitr. I now use knitr to create nearly all of my analysis-summary documents, even those with "brain blob" images, figures, and tables. This post contains a knitr tutorial in the form of an example knitr-created document, and the source needed to recreate it.

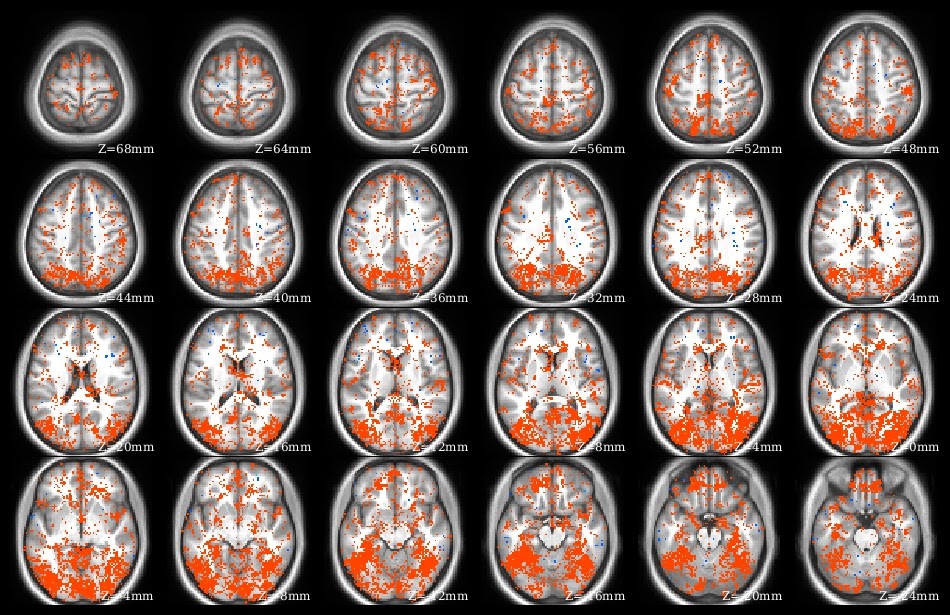
What does knitr do? Yihui has many demonstrations on his web site. I use knitr to create pdf files presenting, summarizing, and interpreting analysis results. Part of the demo pdf is in the image at left to give the idea: I have several paragraphs of explanatory text above a series of overlaid brain images, along with graphs and tables. This entire pdf was created from a knitr .rnw source file, which contains LaTeX text and R code blocks.
Previously, I'd make Word documents describing an analysis, copy-pasting figures and screenshots as needed, and manually formatting tables. Besides time, a big drawback of this system is human memory ... "how exactly did I calculate these figures?." I tried including links to the source R files and notes about thresholds, etc, but often missed some key detail, which I'd then have to reverse-engineer. knitr avoids that problem: I can look at the document's .rnw source code and immediately see which NIfTI image is displayed, which directory contains the plotted data, etc.
In addition to (human) memory and reproducibility benefits, the time saved by using knitr instead of Word for analysis summary documents is substantial. Need to change a parameter and rerun an analysis? With knitr there's no need to spend hours updating the images: just change the file names and parameters in the knitr document and recompile. Similarly, the color scaling or displayed slices can be changed easily.

to run the demo
We successfully tested this demo file on Windows, MacOS, and Ubuntu, always using RStudio, but with whichever LaTeX compiler was recommended for the system.Software-wise, first install RStudio, then install a LaTeX compiler; if you'll only be using LaTeX with R I suggest using TinyTeX. Within R, you'll, need the knitr and oro.nifti packages (plus tinytex, if using).
Now, download the files needed for the demo (listed below). These are mostly the low-resolution NIfTI files I've used in previous tutorials, with a new anatomic underlay image, and the knitr .rnw demo file itself. Put all of the image files into a single directory. When knitr compiles it produces many intermediate files, so it is often best to put each .rnw file into its own directory. For example, put all of the image files into c:/temp/demo/, then brainPlotsDemo.rnw into c:/temp/demo/knitr/.
 Next, open brainPlotsDemo.rnw in RStudio. The RStudio GUI tab menu should look like in the screenshot above, complete with a Compile PDF button. But don't click the button yet! Instead, go through Tools then Global Options in the top RStudio menus to bring up the Options dialog box, as shown here. Click on the Sweave icon, then tell it to Weave Rnw files using knitr (marked with yellow arrow). Then click Ok to close the dialog box, and everything should be ready. In my experience, RStudio just finds the LaTeX installation - you don't need to set the paths yourself.
Next, open brainPlotsDemo.rnw in RStudio. The RStudio GUI tab menu should look like in the screenshot above, complete with a Compile PDF button. But don't click the button yet! Instead, go through Tools then Global Options in the top RStudio menus to bring up the Options dialog box, as shown here. Click on the Sweave icon, then tell it to Weave Rnw files using knitr (marked with yellow arrow). Then click Ok to close the dialog box, and everything should be ready. In my experience, RStudio just finds the LaTeX installation - you don't need to set the paths yourself.
Good luck!
NOTICE: this post was replaced by a newer version 13 March 2020. I will leave this post here, but strongly suggest you follow the new version instead.
UPDATE 15 January 2019: Here is a post with code for plotting surface (gifti) images in knitr (and R).
UPDATE 31 May 2019: Added a copy of the full text of brainPlotsDemo.rnw in below the jump. Note that this includes the make.plots function to plot a row of NIfTI images.
UPDATE 19 July 2019: Added recommendation to use TinyTeX as the LaTeX compiler. After updating R (3.6.0) and RStudio (to 1.2.1335) on my windows computer I was unable to compile to PDF (compilation would start but not complete, leaving empty .synctex(busy) files) with my previous MikTeX setup; switching to TinyTeX fixed the problems. Also slightly edited the demo text and graphs.






















{kind=link}